LIBRETTO-531
LIBRETTO-531: A Phase III superiority trial evaluating Retevmo versus cabozantinib or vandetanib in RET-mutant advanced or metastatic MTC 1,2*†‡
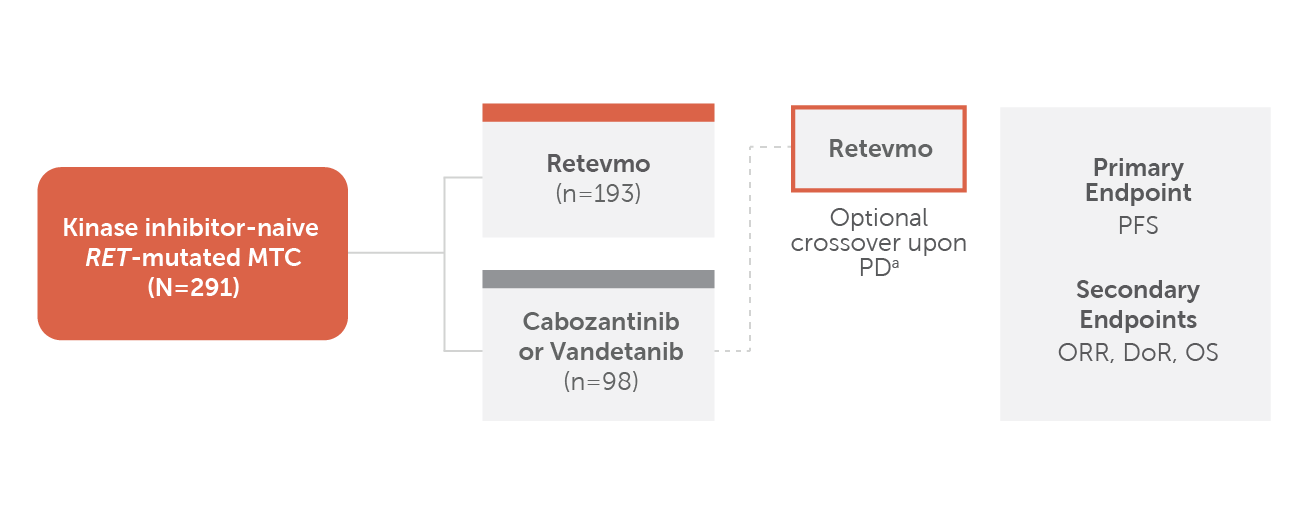
Chart showing trial design for LIBRETTO-531. Patients with kinase inhibitor-naive RET-mutant MTC (N=291) were randomized 2:1. One hundred ninety-three patients were randomized to Retevmo; 98 patients were randomized to cabozantinib or vandetanib. (73 patients and 25 patients received cabozantinib and vandetanib, respectively, within the control arm. Stratification factors: investigator’s choice of treatment if randomized to control arm [choice/intent of treatment regimen must be declared prior to randomization], mutation status [M918T versus other].) Crossover to Retevmo was optional for patients in the control arm upon disease progression (PD) confirmed by IRC. The primary endpoint was PFS. Secondary endpoints included ORR, DoR and OS. (All primary and secondary endpoints listed were reviewed by IRC per RECIST v1.1.)
a PD was confirmed by IRC
- Trial excluded patients who had presence of other validated oncogenic drivers.
- Retevmo was dosed at 160 mg BID. Cabozantinib was dosed at 140 mg QD and vandetanib was dosed at 300 mg QD.
*All primary and secondary endpoints listed were reviewed by IRC per RECIST v1.1.
†Stratification factors: investigator’s choice of treatment if randomized to control arm (choice/intent of treatment regimen must be declared prior to randomization), mutation status (M918T versus other).1
‡73 patients and 25 patients received cabozantinib and vandetanib, respectively, within the control arm. Numerical imbalance may be due to worldwide supply shortage of vandetanib during the trial.2
BID=twice a day; DoR=duration of response; IRC=independent review committee; MTC=medullary thyroid cancer; ORR=overall response rate; PD=progressive disease; PFS=progression-free survival; QD=every day; RECIST=Response Evaluation Criteria in Solid Tumours; RET=rearranged during transfection
PFS
In patients with RET-mutant advanced or metastatic MTC
Retevmo demonstrated superior PFS compared to cabozantinib or vandetanib1,2
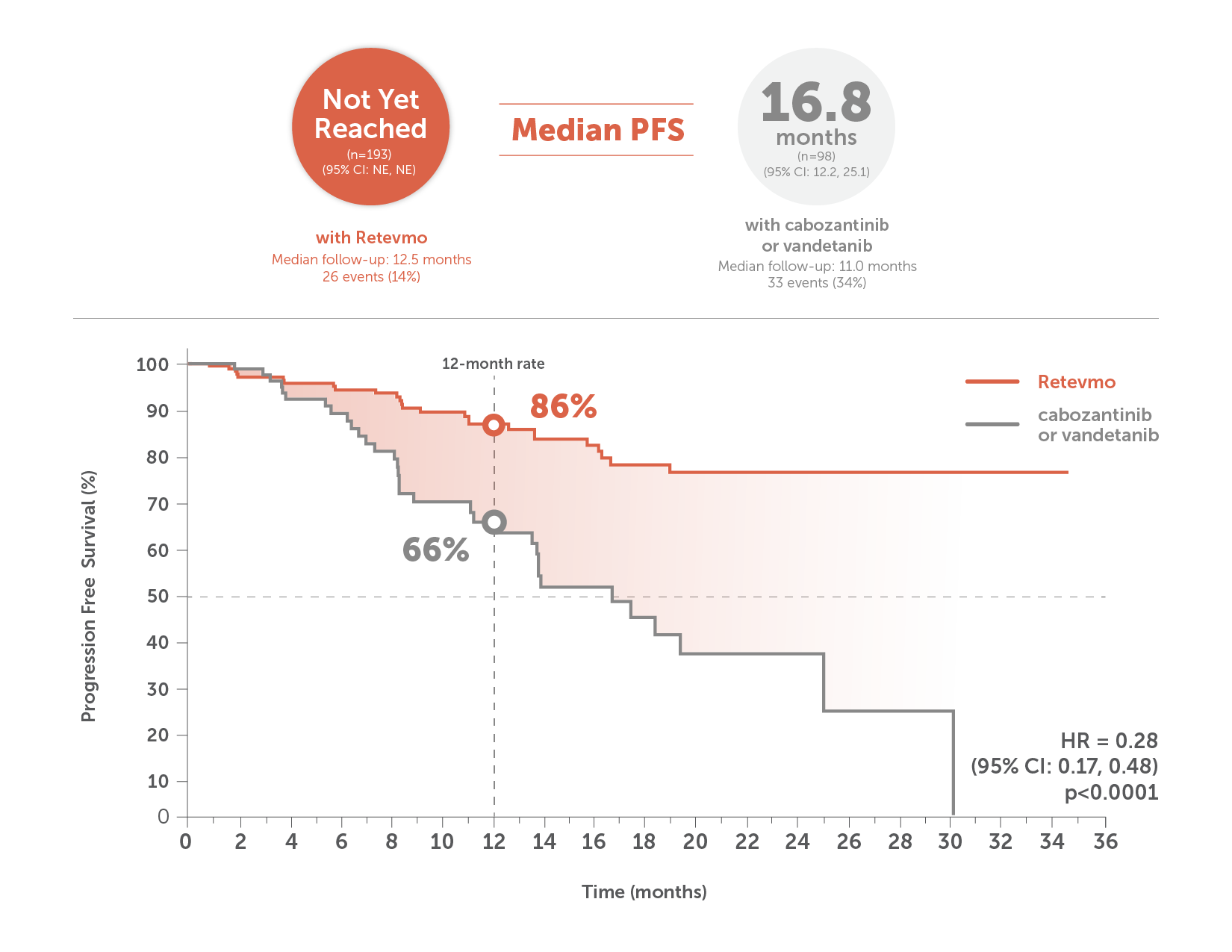
This image shows median PFS in Retevmo-treated patients (n=193) versus those treated with cabozantinib or vandetanib (n=98). PFS was assessed in all randomized patients and was based on an interim analysis with a data cutoff date of May 22, 2023. For patients treated with Retevmo, median PFS was not yet reached (95% CI not estimable) with median follow-up of 12.5 months. In patients treated with cabozantinib or vandetanib, median PFS was 16.8 months (95% CI: 12.2, 25.1) with median follow-up of 11.0 months. An accompanying Kaplan-Meier curve shows progression-free survival for Retevmo-treated patients of 86% at 12 months. PFS for patients treated with cabozantinib or vandetanib was 66% at 12 months. Hazard ratio was 0.28 (p<0.0001).
PFS was assessed in all randomized patients. Based on an interim analysis with a data cutoff date of May 22, 2023.1,2
HR=hazard ratio; NE=not estimable
Select Important Safety Information
Hepatotoxicity: Serious hepatic adverse reactions occurred in 3% of patients treated with Retevmo. Increased aspartate aminotransferase (AST) occurred in 59% of patients, including Grade 3 or 4 events in 11% and increased alanine aminotransferase (ALT) occurred in 55% of patients, including Grade 3 or 4 events in 12%. Monitor ALT and AST prior to initiating Retevmo, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce dose, or permanently discontinue Retevmo based on severity.
ORR and DoR
Overall response rate and duration of response for Retevmo vs cabozantinib or vandetanib1,2,§

Image showing Overall Response Rate for Retevmo versus cabozantinib or vandetanib. ORR for Retevmo (n=193) was 69% (95% CI: 62, 76). Within this group, 58% of patients showed Partial Response and 12% showed Complete Response. In patients treated with cabozantinib or vandetanib (n=98), ORR was 39% (95% CI: 29, 49). Within this group, 35% of patients showed Partial Response and 4% showed Complete Response.

Image showing Duration of Response for Retevmo versus cabozantinib or vandetanib. DoR for Retevmo was not yet reached (95% CI: NE, NE). Median follow-up was 11.1 months. In patients treated with cabozantinib or vandetanib DoR was 16.6 months (95% CI: 10.4, NE). Median follow-up was 12.8 months.
- ORR, a secondary endpoint, was defined as CR+PR and was assessed by the IRC according to RECIST v1.1.
- Based on an interim analysis with a data cutoff date of May 22, 2023.1,2
- At the time of this analysis, overall survival (OS) data were immature with 18 deaths observed.1
§Due to rounding, percentages presented may not add up to the indicated totals.
CI=confidence interval; CR=complete response; PR=partial response
Safety
Safety and tolerability were evaluated for Retevmo in LIBRETTO-5311
Adverse Events (≥10%) in Patients Who Received Retevmo in LIBRETTO-531
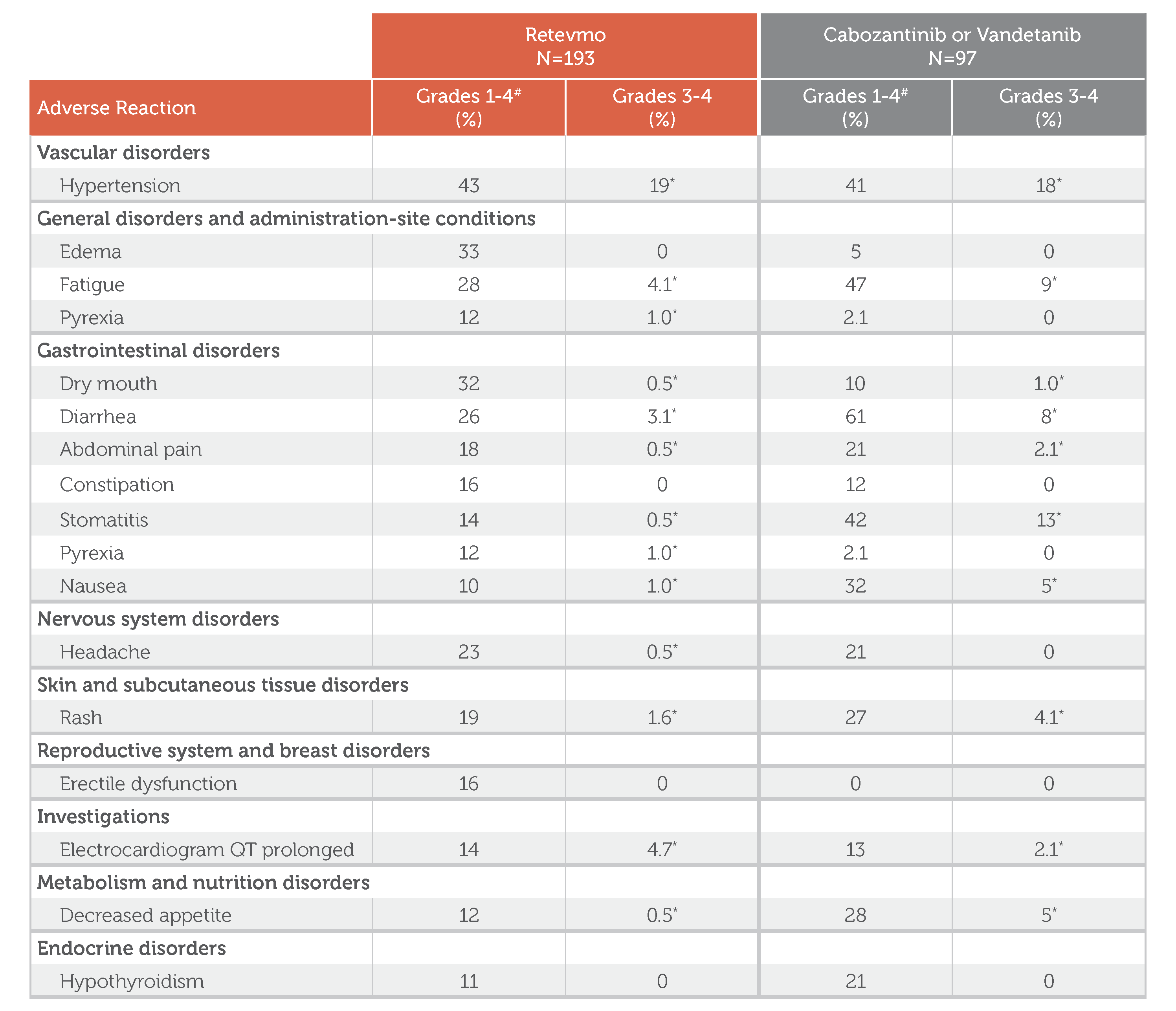
Adverse Events (≥10%) in Patients Who Received Retevmo in LIBRETTO-531
Retevmo, Grades 1-4: In Retevmo-treated patients (n=193), Vascular disorders of Grades 1-4 included hypertension (43%). General disorders and administration-site conditions of Grades 1-4 included edema (33%), fatigue (28%), and pyrexia (12%). Gastrointestinal disorders of Grades 1-4 included dry mouth (32%), diarrhea (26%), abdominal pain (18%), constipation (16%), stomatitis (14%), pyrexia (12%), and nausea (10%). Nervous system disorders of Grades 1-4 included headache (23%). Skin and subcutaneous tissue disorders of Grades 1-4 included rash (19%). Reproductive system and breast disorders of Grades 1-4 included erectile dysfunction (16%). Investigations category included electrocardiogram QT prolonged (14%). Metabolism and nutrition disorders of Grades 1-4 included decreased appetite (12%). Endocrine disorders of Grades 1-4 included hypothyroidism (11%).
Retevmo, Grades 3-4: In Retevmo-treated patients (n=193), Vascular disorders of Grades 3-4 included hypertension (19%). General disorders and administration-site conditions of Grades 3-4 included edema (0.0%), fatigue (4.1%), and pyrexia (1.0%). Gastrointestinal disorders of Grades 3-4 included dry mouth (0.5%), diarrhea (3.1%), abdominal pain (0.5%), constipation (0.0%), stomatitis (0.5%), pyrexia (1.0%), and nausea (1.0%). Nervous system disorders of Grades 3-4 included headache (0.5%). Skin and subcutaneous tissue disorders of Grades 3-4 included rash (1.6%). Reproductive system and breast disorders of Grades 3-4 included erectile dysfunction (0.0%). Investigations category included electrocardiogram QT prolonged (4.7%). Metabolism and nutrition disorders of Grades 3-4 included decreased appetite (0.5%). Endocrine disorders of Grades 3-4 included hypothyroidism (0.0%). All non-zero values for Retevmo Grades 3-4 are footnoted to indicate: “Only includes a Grade 3 adverse reaction.”
Cabozantinib or Vandetanib, Grades 1-4: In cabozantinib- or vandetanib-treated patients (n=97), Vascular disorders of Grades 1-4 included hypertension (41%). General disorders and administration-site conditions of Grades 1-4 included edema (5%), fatigue (47%), and pyrexia (2.1%). Gastrointestinal disorders of Grades 1-4 included dry mouth (10%), diarrhea (61%), abdominal pain (21%), constipation (12%), stomatitis (42%), pyrexia (2.1%), and nausea (32%). Nervous system disorders of Grades 1-4 included headache (21%). Skin and subcutaneous tissue disorders of Grades 1-4 included rash (27%). Reproductive system and breast disorders of Grades 1-4 included erectile dysfunction (0.0%). Investigations category included electrocardiogram QT prolonged (13%). Metabolism and nutrition disorders of Grades 1-4 included decreased appetite (28%). Endocrine disorders of Grades 1-4 included hypothyroidism (21%).
Cabozantinib or Vandetanib, Grades 3-4: In cabozantinib- or vandetanib-treated patients (n=97), Vascular disorders of Grades 3-4 included hypertension (18%). General disorders and administration-site conditions of Grades 3-4 included edema (0.0%), fatigue (9%), and pyrexia (0.0%). Gastrointestinal disorders of Grades 3-4 included dry mouth (1.0%), diarrhea (8%), abdominal pain (2.1%), constipation (0.0%), stomatitis (13%), pyrexia (0.0%), and nausea (5%). Nervous system disorders of Grades 3-4 included headache (0.0%). Skin and subcutaneous tissue disorders of Grades 3-4 included rash (4.1%). Reproductive system and breast disorders of Grades 3-4 included erectile dysfunction (0.0%). Investigations category included electrocardiogram QT prolonged (2.1%). Metabolism and nutrition disorders of Grades 3-4 included decreased appetite (5%). Endocrine disorders of Grades 3-4 included hypothyroidism (0.0%). All non-zero values for Cabozantinib or Vandetanib Grades 3-4 are footnoted to indicate: “Only includes a Grade 3 adverse reaction.”
Footnote: Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 5.0.
*Only includes a Grade 3 adverse reaction
#Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 5.0.
Select Laboratory Abnormalities (≥5%) Worsening from Baseline in Patients Treated with Retevmo in
LIBRETTO-531
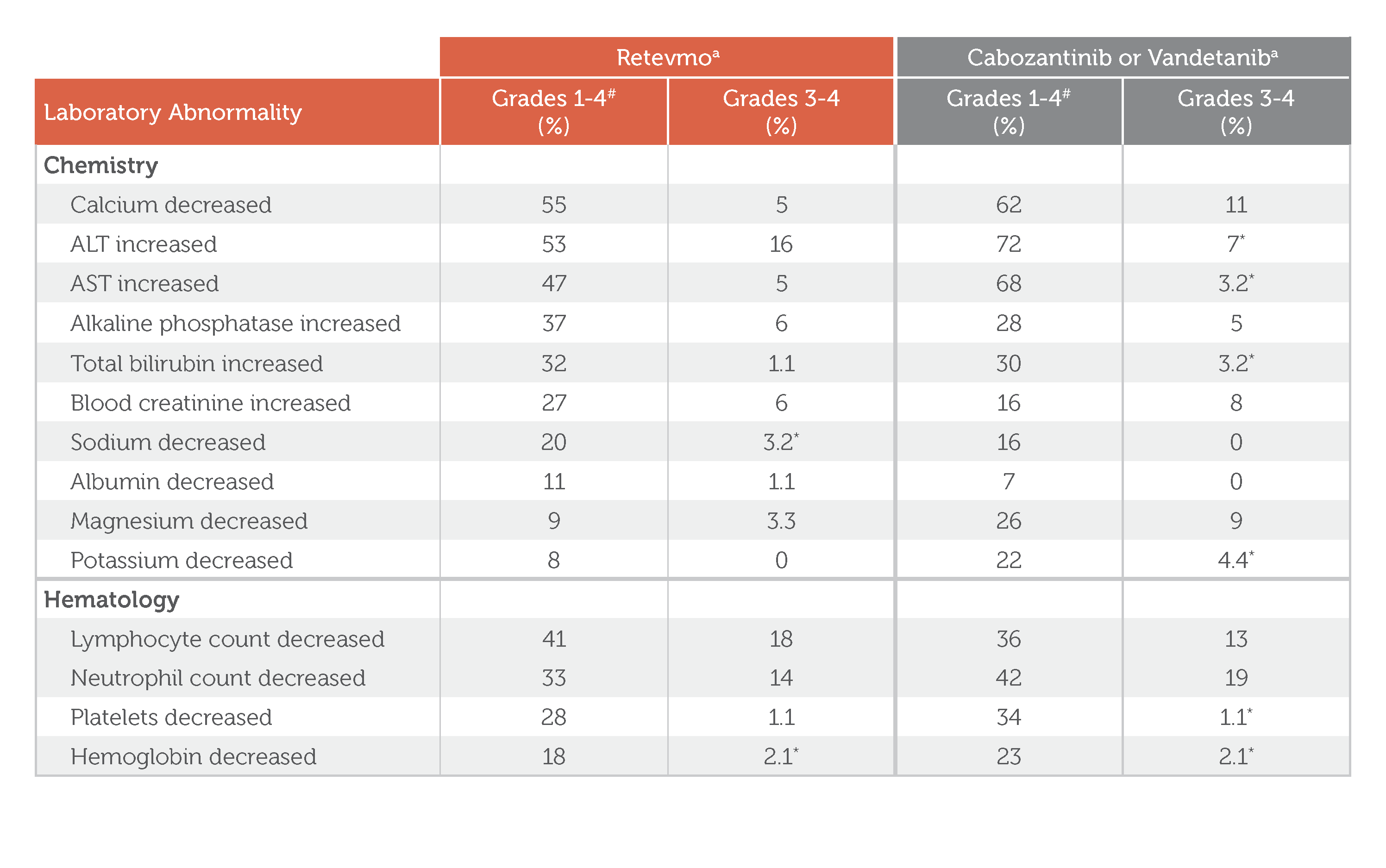
Select Laboratory Abnormalities (≥5%) Worsening from Baseline in Patients Treated with Retevmo in LIBRETTO-531
Retevmo, Grades 1-4: In Retevmo-treated patients, Chemistry-related laboratory abnormalities of Grades 1-4 included calcium decreased (55%), ALT increased (53%), AST increased (47%), alkaline phosphatase increased (37%), total bilirubin increased (32%), blood creatinine increased (27%) sodium decreased (20%), albumin decreased (11%), magnesium decreased (9%), and potassium decreased (8%). Hematology-related laboratory abnormalities of Grades 1-4 included lymphocyte count decreased (41%), neutrophil count decreased (33%), platelets decreased (28%), and hemoglobin decreased (18%).
Retevmo, Grades 3-4: In Retevmo-treated patients, Chemistry-related laboratory abnormalities of Grades 3-4 included calcium decreased (5%), ALT increased (16%), AST increased (5%), alkaline phosphatase increased (6%), total bilirubin increased (1.1%), blood creatinine increased (6%) sodium decreased (3.2%; Grade 3 AE only), albumin decreased (1.1%), magnesium decreased (3.3%), and potassium decreased (0.0%). Hematology-related laboratory abnormalities of Grades 3-4 included lymphocyte count decreased (18%), neutrophil count decreased (14%), platelets decreased (1.1%), and hemoglobin decreased (2.1%; Grade 3 AE only).
Cabozantinib or Vandetanib, Grades 1-4: In cabozantinib- or vandetanib-treated patients, Chemistry-related laboratory abnormalities of Grades 1-4 included calcium decreased (62%), ALT increased (72%), AST increased (68%), alkaline phosphatase increased (28%), total bilirubin increased (30%), blood creatinine increased (16%) sodium decreased (16%), albumin decreased (7%), magnesium decreased (26%), and potassium decreased (22%). Hematology-related laboratory abnormalities of Grades 1-4 included lymphocyte count decreased (36%), neutrophil count decreased (42%), platelets decreased (34%), and hemoglobin decreased (23%).
Cabozantinib or Vandetanib, Grades 3-4: In cabozantinib- or vandetanib-treated patients, Chemistry-related laboratory abnormalities of Grades 3-4 included calcium decreased (11%), ALT increased (7%; Grade 3 AE only), AST increased (3.2%), alkaline phosphatase increased (5%), total bilirubin increased (3.2%; Grade 3 AE only), blood creatinine increased (8%) sodium decreased (0.0%), albumin decreased (0.0%), magnesium decreased (9%), and potassium decreased (4.4%; Grade 3 AE only). Hematology-related laboratory abnormalities of Grades 3-4 included lymphocyte count decreased (13%), neutrophil count decreased (19%), platelets decreased (1.1%; Grade 3 AE only), and hemoglobin decreased (2.1%; Grade 3 AE only).
Treatment group footnote: Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available: Retevmo (range: 183 to 191 patients) and chemotherapy with or without cabozantinib or vandetanib (range: 91 to 94 patients).
a Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available: Retevmo (range: 183 to 191 patients) and chemotherapy with or without cabozantinib or vandetanib (range: 91 to 94 patients).1
- Serious adverse reactions occurred in 22% of patients who received Retevmo. The most frequent serious adverse reactions were pneumonia and pyrexia (n = 3, each) and hypertension and urinary tract infection (n = 2, each).
- Fatal adverse reactions occurred in 2.1% of patients; fatal adverse reactions included COVID-19, diabetic ketoacidosis, multiple organ dysfunction syndrome, and sudden death (n=1 each).
- Permanent discontinuation due to an adverse reaction occurred in 4.7% of patients who received Retevmo. Adverse reactions resulting in permanent discontinuation were edema, multiple organ dysfunction syndrome, sudden death, and AST increased, diabetic ketoacidosis, chronic kidney disease, retinopathy, COVID-19 and somatic symptom disorder (n = 1, each).
- Dosage interruptions due to an adverse reaction occurred in 49% of patients who received Retevmo. Adverse reactions requiring dosage omission in ≥5% of patients included ALT increased (9%) and hypertension (7%).
- Dose reductions due to an adverse reaction occurred in 39% of patients who received Retevmo. One adverse reaction, increased ALT (7%), required a dose reduction in ≥5% of patients.
- The most common adverse reactions (≥25%) in patients who received Retevmo were hypertension, edema, dry mouth, fatigue, diarrhea.
- The most common Grade 3 or 4 laboratory abnormalities (≥5%) in patients who received Retevmo were decreased lymphocyte, increased ALT, decreased neutrophils, increased ALP, increased blood creatinine, decreased calcium, and increased AST.
- Clinically relevant adverse reactions in ≤10% of patients who received Retevmo include dizziness (8%); urinary tract infections (8%); vomiting (8%); pneumonia, interstitial lung disease/pneumonitis, chylous ascites and hypersensitivity (all < 2%).
For complete details on adverse reactions and laboratory abnormalities, please see full Prescribing Information.
AE=adverse event; ALT=alanine transaminase; AST=aspartate aminotransferase; COVID-19=coronavirus disease 2019; GGT=gamma-glutamyl transferase; INR=international normalized ratio; LFT=liver function test; WBC=white blood cell
Select Important Safety Information
Severe, life-threatening, and fatal interstitial lung disease (ILD)/pneumonitis can occur in patients treated with Retevmo. ILD/pneumonitis occurred in 1.8% of patients who received Retevmo, including 0.3% with Grade 3 or 4 events, and 0.3% with fatal reactions. Monitor for pulmonary symptoms indicative of ILD/pneumonitis. Withhold Retevmo and promptly investigate for ILD in any patient who presents with acute or worsening of respiratory symptoms which may be indicative of ILD (e.g., dyspnea, cough, and fever). Withhold, reduce dose, or permanently discontinue Retevmo based on severity of confirmed ILD.
L-001 Efficacy Summary
In patients with RET-mutant advanced or metastatic MTC
Retevmo was granted initial approval based on LIBRETTO-001, a Phase I/II trial
Efficacy in cabozantinib/vandetanib-naive patients (n=88)1,3

Graphic showing Objective Response Rate and Median Duration of Response for Retevmo treatment in cabozantinib/vandetanib-naive patients. ORR was 81% (95% CI: 71, 88), with 28% complete response (CR) and 52% PR (partial response). Median DoR not yet reached (95% CI: 51.3, NE). Median follow-up was 44.2 months. Due to rounding, percentages presented may not add up to the indicated totals.
Major efficacy outcome measures for patients previously treatedǁ with cabozantinib and/or vandetanib (n=55)1:
-
ORR = 76% (95% CI: 63, 87)
- 18% CR + 58% PR
- Median DoR = 45.3 months (95% CI: 29.9, NE); Median follow-up: 50.7 months3
Time-to-event endpoints are not interpretable in a single-arm study. The clinical significance of this descriptive analysis is not known.
ORR was defined as CR + PR and was assessed by IRC according to RECIST v1.1.1
All results reviewed by an IRC.1
Due to rounding, percentages presented may not add up to the indicated totals.
ǁThe efficacy of Retevmo was evaluated in 55 patients with RET-mutant advanced MTC who were previously treated with cabozantinib or vandetanib enrolled into a cohort of LIBRETTO-001.1
L-001 Trial Design
The phase I/II, multicohort, open-label, single-arm, multicenter LIBRETTO-001 trial included a pooled safety population of 796 patients with locally advanced or metastatic RET fusion-positive NSCLC¶, advanced or metastatic RET fusion-positive thyroid cancer (non-MTC)#, advanced or metastatic RET-mutant MTC, and other cancers, including patients with RET alterations in other tumors**. Major efficacy outcomes were ORR and DoR, and were evaluated in 565 patients. Other efficacy outcomes, evaluated in subsets of patients, included CNS ORR, CNS DoR, PFS, OS, time to response, and best change in tumor size from baseline. In phase II, the dose for Retevmo was 160 mg PO BID.1,4
¶Patients with locally advanced or metastatic RET fusion-positive NSCLC who had progressed on platinum-based chemotherapy and those without prior systemic therapy were enrolled in separate cohorts.1
#Non-MTC by histology included papillary (n=54), poorly differentiated (n=6), anaplastic (n=4), and Hurthle cell (n=1).1
**Other tumors included pancreatic adenocarcinoma (n=11), colorectal cancer (n=10), and salivary cancer (n=4).1
CNS=central nervous system; PO=orally.
Select Important Safety Information
Hypertension occurred in 41% of patients, including Grade 3 hypertension in 20% and Grade 4 in one (0.1%) patient. Overall, 6.3% had their dose interrupted and 1.3% had their dose reduced for hypertension. Treatment-emergent hypertension was most commonly managed with anti-hypertension medications. Do not initiate Retevmo in patients with uncontrolled hypertension. Optimize blood pressure prior to initiating Retevmo. Monitor blood pressure after 1 week, at least monthly thereafter, and as clinically indicated. Initiate or adjust anti-hypertensive therapy as appropriate. Withhold, reduce dose, or permanently discontinue Retevmo based on severity.
References: 1. Retevmo (selpercatinib). Prescribing Information. Lilly USA, LLC. 2. Hadoux J, Elisei R, Brose MS, et al. Phase 3 trial of selpercatinib in advanced RET-mutant medullary thyroid cancer. N Engl J Med. 2023;389(20):1851-1861. doi:10.1056/NEJMoa2309719 3. Data on File, Lilly USA, LLC. DOF-SE-US-0089. 4. Phase 1/2 study of LOXO-292 in patients with advanced solid tumors, RET fusion-positive solid tumors, and medullary thyroid cancer (LIBRETTO-001). https://clinicaltrials.gov/ct2/show/NCT03157128. Updated June 9, 2022. Accessed June 14, 2022.
INDICATIONS
Retevmo is a kinase inhibitor indicated for the treatment of:
- adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with a rearranged during transfection (RET) gene fusion, as detected by an FDA-approved test
- adult and pediatric patients 2 years of age and older with advanced or metastatic medullary thyroid cancer (MTC) with a RET mutation, as detected by an FDA-approved test, who require systemic therapy
- adult and pediatric patients 2 years of age and older with advanced or metastatic thyroid cancer with a RET gene fusion, as detected by an FDA-approved test, who require systemic therapy and who are radioactive iodine-refractory (if radioactive iodine is appropriate)
- adult and pediatric patients 2 years of age and older with locally advanced or metastatic solid tumors with a RET gene fusion, as detected by an FDA-approved test, that have progressed on or following prior systemic treatment or who have no satisfactory alternative treatment options*
*This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DoR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.